 Ευαγγελάτος Γεώργιος")
Το πολύ γνωστό περιοδικό Nature Medicine διαφωνεί, ο κορωνοϊός είναι αποτέλεσμα μεταλλάξεων από ζώο (νυχτερίδα) και μόλυνσης του ανθρώπου παρά τεχνητής τροποποίησης.
The proximal origin of SARS-CoV-2
Nature Medicine volume 26, pages 450–452 (2020)
To the Editor — Since the first reports of novel pneumonia (COVID-19) in Wuhan, Hubei province, China1,2, there has been considerable discussion on the origin of the causative virus, SARS-CoV-23 (also referred to as HCoV-19)4. Infections with SARS-CoV-2 are now widespread, and as of 11 March 2020, 121,564 cases have been confirmed in more than 110 countries, with 4,373 deaths5.
SARS-CoV-2 is the seventh coronavirus known to infect humans; SARS-CoV, MERS-CoV and SARS-CoV-2 can cause severe disease, whereas HKU1, NL63, OC43 and 229E are associated with mild symptoms6. Here we review what can be deduced about the origin of SARS-CoV-2 from comparative analysis of genomic data. We offer a perspective on the notable features of the SARS-CoV-2 genome and discuss scenarios by which they could have arisen. Our analyses clearly show that SARS-CoV-2 is not a laboratory construct or a purposefully manipulated virus.
Notable features of the SARS-CoV-2 genome
Our comparison of alpha- and betacoronaviruses identifies two notable genomic features of SARS-CoV-2: (i) on the basis of structural studies7,8,9 and biochemical experiments1,9,10, SARS-CoV-2 appears to be optimized for binding to the human receptor ACE2; and (ii) the spike protein of SARS-CoV-2 has a functional polybasic (furin) cleavage site at the S1–S2 boundary through the insertion of 12 nucleotides8, which additionally led to the predicted acquisition of three O-linked glycans around the site.
1. Mutations in the receptor-binding domain of SARS-CoV-2
The receptor-binding domain (RBD) in the spike protein is the most variable part of the coronavirus genome1,2. Six RBD amino acids have been shown to be critical for binding to ACE2 receptors and for determining the host range of SARS-CoV-like viruses7. With coordinates based on SARS-CoV, they are Y442, L472, N479, D480, T487 and Y4911, which correspond to L455, F486, Q493, S494, N501 and Y505 in SARS-CoV-27. Five of these six residues differ between SARS-CoV-2 and SARS-CoV (Fig. 1a). On the basis of structural studies7,8,9 and biochemical experiments1,9,10, SARS-CoV-2 seems to have an RBD that binds with high affinity to ACE2 from humans, ferrets, (νυφίτσες), cats and other species with high receptor homology7.
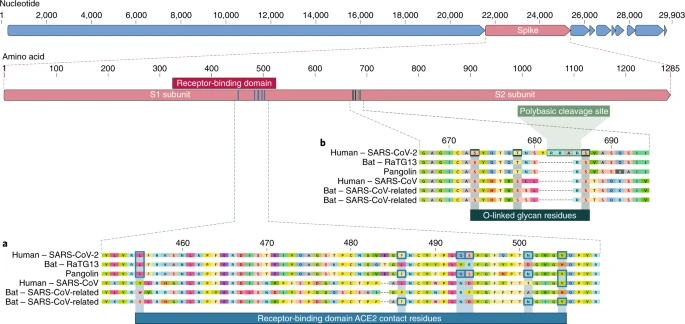
a, Mutations in contact residues of the SARS-CoV-2 spike protein. The spike protein of SARS-CoV-2 (red bar at top) was aligned against the most closely related SARS-CoV-like coronaviruses and SARS-CoV itself. Key residues in the spike protein that make contact to the ACE2 receptor are marked with blue boxes in both SARS-CoV-2 and related viruses, including SARS-CoV (Urbani strain). b, Acquisition of polybasic cleavage site and O-linked glycans. Both the polybasic cleavage site and the three adjacent predicted O-linked glycans are unique to SARS-CoV-2 and were not previously seen in lineage B betacoronaviruses. Sequences shown are from NCBI GenBank, accession codes MN908947, MN996532, AY278741, KY417146 and MK211376. The pangolin coronavirus sequences are a consensus generated from SRR10168377 and SRR10168378 (NCBI BioProject PRJNA573298)29,30.
While the analyses above suggest that SARS-CoV-2 may bind human ACE2 with high affinity, computational analyses predict that the interaction is not ideal7 and that the RBD sequence is different from those shown in SARS-CoV to be optimal for receptor binding7,11. Thus, the high-affinity binding of the SARS-CoV-2 spike protein to human ACE2 is most likely the result of natural selection on a human or human-like ACE2 that permits another optimal binding solution to arise. This is strong evidence that SARS-CoV-2 is not the product of purposeful manipulation.
2. Polybasic furin cleavage site and O-linked glycans
The second notable feature of SARS-CoV-2 is a polybasic cleavage site (RRAR) at the junction of S1 and S2, the two subunits of the spike8 (Fig. 1b). This allows effective cleavage by furin and other proteases and has a role in determining viral infectivity and host range12. In addition, a leading proline is also inserted at this site in SARS-CoV-2; thus, the inserted sequence is PRRA (Fig. 1b). The turn created by the proline is predicted to result in the addition of O-linked glycans to S673, T678 and S686, which flank the cleavage site and are unique to SARS-CoV-2 (Fig. 1b). Polybasic cleavage sites have not been observed in related ‘lineage B’ betacoronaviruses, although other human betacoronaviruses, including HKU1 (lineage A), have those sites and predicted O-linked glycans13. Given the level of genetic variation in the spike, it is likely that SARS-CoV-2-like viruses with partial or full polybasic cleavage sites will be discovered in other species.
The functional consequence of the polybasic cleavage site in SARS-CoV-2 is unknown, and it will be important to determine its impact on transmissibility and pathogenesis in animal models. Experiments with SARS-CoV have shown that insertion of a furin cleavage site at the S1–S2 junction enhances cell–cell fusion without affecting viral entry14. In addition, efficient cleavage of the MERS-CoV spike enables MERS-like coronaviruses from bats to infect human cells15. In avian influenza viruses, rapid replication and transmission in highly dense chicken populations selects for the acquisition of polybasic cleavage sites in the hemagglutinin (HA) protein16, which serves a function similar to that of the coronavirus spike protein. Acquisition of polybasic cleavage sites in HA, by insertion or recombination, converts low-pathogenicity avian influenza viruses into highly pathogenic forms16. The acquisition of polybasic cleavage sites by HA has also been observed after repeated passage in cell culture or through animals17.
The function of the predicted O-linked glycans is unclear, but they could create a ‘mucin-like domain’ that shields epitopes or key residues on the SARS-CoV-2 spike protein18. Several viruses utilize mucin-like domains as glycan shields involved immunoevasion18. Although prediction of O-linked glycosylation is robust, experimental studies are needed to determine if these sites are used in SARS-CoV-2.
Theories of SARS-CoV-2 origins
It is improbable that SARS-CoV-2 emerged through laboratory manipulation of a related SARS-CoV-like coronavirus. As noted above, the RBD of SARS-CoV-2 is optimized for binding to human ACE2 with an efficient solution different from those previously predicted7,11. Furthermore, if genetic manipulation had been performed, one of the several reverse-genetic systems available for betacoronaviruses would probably have been used19. However, the genetic data irrefutably show that SARS-CoV-2 is not derived from any previously used virus backbone20. Instead, we propose two scenarios that can plausibly explain the origin of SARS-CoV-2: (i) natural selection in an animal host before zoonotic transfer; and (ii) natural selection in humans following zoonotic transfer. We also discuss whether selection during passage could have given rise to SARS-CoV-2.
1. Natural selection in an animal host before zoonotic transfer
As many early cases of COVID-19 were linked to the Huanan market in Wuhan1,2, it is possible that an animal source was present at this location. Given the similarity of SARS-CoV-2 to bat SARS-CoV-like coronaviruses2, it is likely that bats serve as reservoir hosts for its progenitor. Although RaTG13, sampled from a Rhinolophus affinis bat1, is ~96% identical overall to SARS-CoV-2, its spike diverges in the RBD, which suggests that it may not bind efficiently to human ACE27 (Fig. 1a).
Malayan pangolins (Manis javanica) illegally imported into Guangdong province contain coronaviruses similar to SARS-CoV-221. Although the RaTG13 bat virus remains the closest to SARS-CoV-2 across the genome1, some pangolin coronaviruses exhibit strong similarity to SARS-CoV-2 in the RBD, including all six key RBD residues21 (Fig. 1). This clearly shows that the SARS-CoV-2 spike protein optimized for binding to human-like ACE2 is the result of natural selection.
Neither the bat betacoronaviruses nor the pangolin betacoronaviruses sampled thus far have polybasic cleavage sites. Although no animal coronavirus has been identified that is sufficiently similar to have served as the direct progenitor of SARS-CoV-2, the diversity of coronaviruses in bats and other species is massively undersampled. Mutations, insertions and deletions can occur near the S1–S2 junction of coronaviruses22, which shows that the polybasic cleavage site can arise by a natural evolutionary process. For a precursor virus to acquire both the polybasic cleavage site and mutations in the spike protein suitable for binding to human ACE2, an animal host would probably have to have a high population density (to allow natural selection to proceed efficiently) and an ACE2-encoding gene that is similar to the human ortholog.
2. Natural selection in humans following zoonotic transfer
It is possible that a progenitor of SARS-CoV-2 jumped into humans, acquiring the genomic features described above through adaptation during undetected human-to-human transmission. Once acquired, these adaptations would enable the pandemic to take off and produce a sufficiently large cluster of cases to trigger the surveillance system that detected it1,2.
All SARS-CoV-2 genomes sequenced so far have the genomic features described above and are thus derived from a common ancestor that had them too. The presence in pangolins of an RBD very similar to that of SARS-CoV-2 means that we can infer this was also probably in the virus that jumped to humans. This leaves the insertion of polybasic cleavage site to occur during human-to-human transmission.
Estimates of the timing of the most recent common ancestor of SARS-CoV-2 made with current sequence data point to emergence of the virus in late November 2019 to early December 201923, compatible with the earliest retrospectively confirmed cases24. Hence, this scenario presumes a period of unrecognized transmission in humans between the initial zoonotic event and the acquisition of the polybasic cleavage site. Sufficient opportunity could have arisen if there had been many prior zoonotic events that produced short chains of human-to-human transmission over an extended period. This is essentially the situation for MERS-CoV, for which all human cases are the result of repeated jumps of the virus from dromedary camels, producing single infections or short transmission chains that eventually resolve, with no adaptation to sustained transmission25.
Studies of banked human samples could provide information on whether such cryptic spread has occurred. Retrospective serological studies could also be informative, and a few such studies have been conducted showing low-level exposures to SARS-CoV-like coronaviruses in certain areas of China26. Critically, however, these studies could not have distinguished whether exposures were due to prior infections with SARS-CoV, SARS-CoV-2 or other SARS-CoV-like coronaviruses. Further serological studies should be conducted to determine the extent of prior human exposure to SARS-CoV-2.
3. Selection during passage
Basic research involving passage of bat SARS-CoV-like coronaviruses in cell culture and/or animal models has been ongoing for many years in biosafety level 2 laboratories across the world27, and there are documented instances of laboratory escapes of SARS-CoV28. We must therefore examine the possibility of an inadvertent laboratory release of SARS-CoV-2.
In theory, it is possible that SARS-CoV-2 acquired RBD mutations (Fig. 1a) during adaptation to passage in cell culture, as has been observed in studies of SARS-CoV11. The finding of SARS-CoV-like coronaviruses from pangolins with nearly identical RBDs, however, provides a much stronger and more parsimonious explanation of how SARS-CoV-2 acquired these via recombination or mutation19.
The acquisition of both the polybasic cleavage site and predicted O-linked glycans also argues against culture-based scenarios. New polybasic cleavage sites have been observed only after prolonged passage of low-pathogenicity avian influenza virus in vitro or in vivo17. Furthermore, a hypothetical generation of SARS-CoV-2 by cell culture or animal passage would have required prior isolation of a progenitor virus with very high genetic similarity, which has not been described. Subsequent generation of a polybasic cleavage site would have then required repeated passage in cell culture or animals with ACE2 receptors similar to those of humans, but such work has also not previously been described. Finally, the generation of the predicted O-linked glycans is also unlikely to have occurred due to cell-culture passage, as such features suggest the involvement of an immune system18.
Conclusions
In the midst of the global COVID-19 public-health emergency, it is reasonable to wonder why the origins of the pandemic matter. Detailed understanding of how an animal virus jumped species boundaries to infect humans so productively will help in the prevention of future zoonotic events. For example, if SARS-CoV-2 pre-adapted in another animal species, then there is the risk of future re-emergence events. In contrast, if the adaptive process occurred in humans, then even if repeated zoonotic transfers occur, they are unlikely to take off without the same series of mutations. In addition, identifying the closest viral relatives of SARS-CoV-2 circulating in animals will greatly assist studies of viral function. Indeed, the availability of the RaTG13 bat sequence helped reveal key RBD mutations and the polybasic cleavage site.
The genomic features described here may explain in part the infectiousness and transmissibility of SARS-CoV-2 in humans. Although the evidence shows that SARS-CoV-2 is not a purposefully manipulated virus, it is currently impossible to prove or disprove the other theories of its origin described here. However, since we observed all notable SARS-CoV-2 features, including the optimized RBD and polybasic cleavage site, in related coronaviruses in nature, we do not believe that any type of laboratory-based scenario is plausible.
More scientific data could swing the balance of evidence to favor one hypothesis over another. Obtaining related viral sequences from animal sources would be the most definitive way of revealing viral origins. For example, a future observation of an intermediate or fully formed polybasic cleavage site in a SARS-CoV-2-like virus from animals would lend even further support to the natural-selection hypotheses. It would also be helpful to obtain more genetic and functional data about SARS-CoV-2, including animal studies. The identification of a potential intermediate host of SARS-CoV-2, as well as sequencing of the virus from very early cases, would similarly be highly informative. Irrespective of the exact mechanisms by which SARS-CoV-2 originated via natural selection, the ongoing surveillance of pneumonia in humans and other animals is clearly of utmost importance.
References
- 1.
Zhou, P. et al. Nature https://doi.org/10.1038/s41586-020-2012-7 (2020).
- 2.
Wu, F. et al. Nature https://doi.org/10.1038/s41586-020-2008-3 (2020).
- 3.
Gorbalenya, A. E. et al. bioRxiv https://doi.org/10.1101/2020.02.07.937862 (2020).
- 4.
Jiang, S. et al. Lancet https://doi.org/10.1016/S0140-6736(20)30419-0 (2020).
- 5.
Dong, E., Du, H. & Gardner, L. Lancet Infect. Dis. https://doi.org/10.1016/S1473-3099(20)30120-1 (2020).
- 6.
Corman, V. M., Muth, D., Niemeyer, D. & Drosten, C. Adv. Virus Res. 100, 163–188 (2018).
- 7.
Wan, Y., Shang, J., Graham, R., Baric, R. S. & Li, F. J. Virol. https://doi.org/10.1128/JVI.00127-20 (2020).
- 8.
Walls, A. C. et al. bioRxiv https://doi.org/10.1101/2020.02.19.956581 (2020).
- 9.
Wrapp, D. et al. Science https://doi.org/10.1126/science.abb2507 (2020).
- 10.
Letko, M., Marzi, A. & Munster, V. Nat. Microbiol. https://doi.org/10.1038/s41564-020-0688-y (2020).
- 11.
Sheahan, T. et al. J. Virol. 82, 2274–2285 (2008).
- 12.
Nao, N. et al. MBio 8, e02298-16 (2017).
- 13.
Chan, C.-M. et al. Exp. Biol. Med. 233, 1527–1536 (2008).
- 14.
Follis, K. E., York, J. & Nunberg, J. H. Virology 350, 358–369 (2006).
- 15.
Menachery, V. D. et al. J. Virol. https://doi.org/10.1128/JVI.01774-19 (2019).
- 16.
Alexander, D. J. & Brown, I. H. Rev. Sci. Tech. 28, 19–38 (2009).
- 17.
Ito, T. et al. J. Virol. 75, 4439–4443 (2001).
- 18.
Bagdonaite, I. & Wandall, H. H. Glycobiology 28, 443–467 (2018).
- 19.
Cui, J., Li, F. & Shi, Z.-L. Nat. Rev. Microbiol. 17, 181–192 (2019).
- 20.
Almazán, F. et al. Virus Res. 189, 262–270 (2014).
- 21.
Zhang, T., Wu, Q. & Zhang, Z. bioRxiv https://doi.org/10.1101/2020.02.19.950253 (2020).
- 22.
Yamada, Y. & Liu, D. X. J. Virol. 83, 8744–8758 (2009).
- 23.
Rambaut, A. Virological.org http://virological.org/t/356 (2020).
- 24.
Huang, C. et al. Lancet https://doi.org/10.1016/S0140-6736(20)30183-5 (2020).
- 25.
Dudas, G., Carvalho, L. M., Rambaut, A. & Bedford, T. eLife 7, e31257 (2018).
- 26.
Wang, N. et al. Virol. Sin. 33, 104–107 (2018).
- 27.
Ge, X.-Y. et al. Nature 503, 535–538 (2013).
- 28.
Lim, P. L. et al. N. Engl. J. Med. 350, 1740–1745 (2004).
- 29.
Wong, M. C., Javornik Cregeen, S. J., Ajami, N. J. & Petrosino, J. F. bioRxiv https://doi.org/10.1101/2020.02.07.939207 (2020).
- 30.
Liu, P., Chen, W. & Chen, J.-P. Viruses 11, 979 (2019).
Acknowledgements
We thank all those who have contributed sequences to the GISAID database (https://www.gisaid.org/) and analyses to Virological.org (http://virological.org/). We thank M. Farzan for discussions, and the Wellcome Trust for support. K.G.A. is a Pew Biomedical Scholar and is supported by NIH grant U19AI135995. A.R. is supported by the Wellcome Trust (Collaborators Award 206298/Z/17/Z―ARTIC network) and the European Research Council (grant agreement no. 725422―ReservoirDOCS). E.C.H. is supported by an ARC Australian Laureate Fellowship (FL170100022). R.F.G. is supported by NIH grants U19AI135995, U54 HG007480 and U19AI142790.
Ethics declarations
Competing interests
R.F.G. is co-founder of Zalgen Labs, a biotechnology company that develops countermeasures to emerging viruses.
Rights and permissions
Σ.γ.: Με το πιο πάνω άρθρο όλα είναι τακτοποιημένα, οι κυβερνήσεις μπορεί να το χρησιμοποιούν για το καλό μας, οι γιατροί για να δικαιολογούν τον εμβολιασμό όλων μας για να “σωθούμε” και όσοι πεθαίνουν από τις παρενέργειες του “τσιμπήματος”, αυτό ¨οφείλεται σε άλλες αιτίες” πλην των εμβολίων. Δείτε την παρακάτω ανάρτηση:
Πρόσφατα Σχόλια